Lab 4a, 4b, 4i, 4j-DNA
Materials for Lab 4
Analytical balance
Tabletop milligram balance
7.6 cm x 7.6 cm weigh paper
3.5 inches x 3.5 inches weigh boat
Lab scoops
Sodium chloride
Tubes, 15 mL, capped
Tube racks for 15 mL tubes
TRIS
EDTA, disodium salt
Bottle, 125 mL
Graduated cylinder, 100 mL
Ph paper
Hydrochloric acid
Sodium hydroxide
Glass rods
50 mL beakers
Salmon sperm sample
2mL pipet
Micropipet, P-1000
95% Ethanol
Sharpie
Plastic beaker 1L tripour
40x TAE buffer concentrate
600 mL beaker
Agarose
250 mL Media bottle
Microwave Oven
Hot hands protector
Horizontal gel box
65 degree Celsius water bath
Prepared agarose gel
Tube rack for 1.7 mL tubes
Reaction tubes, 1.7 mL
DNA samples
Yeast DNA 50 micrometers (g)/micrometer(liter) and loading dye
pBR322 50 micrometers (g)/micrometer(liter) and dye
Lambda 50 micrometers (g)/micrometer(liter) and loading dye
Other loading samples and dye
Gel loading dye 10X or 6X
Micropipet, P-10
Micropipet, P-100
Micropipet tips for P-100
Microcentrifuge
Lambda/ HimIII, 50 micrometers (g)/micrometer(liter) and dye
power supply
Ethidium bromide, 0.5 micrometer(g)/ mL
Gel photo imaging system
Thermal paper
Thermal paper
Large gloves
Safety plastic glasses
5.5 inches x 5.5 inches weigh boat
Lab 4a and 4b
Why NaCl? Na+ bind DNA so it can clump together and can precipitate
Precipitation- taking something out of a solution
DNases- breaks down DNA
Why Tris? buffer, maintains pH
why EDTA? prevents DNA braked down negative binds ca++ and mg++ co-factors enzymes. Prevents DNases activity
Purpose:
to make 10 ml of 5m NaCl
100mL of 10mM Tris, 1mM EDTA
M= mole/L
mM= 1/1000 mole/L
1 mole= 6.02 x 10^23 particles
Molarity Calculations
g substance needed=(M)(L)(g/mol)
Solution 1:
5M NaCl = 10mL
1. 10mL 5M NaCl
(5M) (0.01L) (58.44 g/mol) = 2.92 g NaCl NaCl-formula weight = 58.44 g/mol
2. TE
Tris (0.01M)(0.1L)(157.6 g/mol) = 0.158 g Tris
EDTA (0.001M)(0.1L)(372.24 g/mol) = 0.0372 g EDTA
Label
What it is- NaCl
Concentration- 5M desired ph 7.5-8.5 current ph is 6.3-6.5 so need to add base(NaCl)
date- 10-9-14
initials- NR, ZM, JM, RS,MI
period- 2/3
C1V1 (stock solution concentration)=C2V2 (final desired concentration)= final desired volume
C1= volume of stock solution to use V1= C2V2/C1
C=4 mg/ml
V1= ?
C2= 2 mg/ml
V2= 2 mL
Final ph 7.7
Purpose 4b was to spool DNA to extract from liquid.
1. Dilute DNA w/TE -1 mL DNA
1m beaker,observe - 1 mL TE
2. Add NaCl
3. Add 4 mL EtoH(trickle down side) observe
4. Spool DNA
5. Put DNA into new tube w/ 2 mL fresh Te and Label:
Observations
1. The substance is transparent and somewhat congealed.
2. The substance is still clear but there is a different layer and the DNA is showing.
3. The DNA is dense and is more right than the rest of the substance.
4. DNA was kind of viscus and goopy. Me and my partner had similar results to everyone in class.
We left no DNA in beaker. You need to know howto extract DNA so you can study it by finding sequence of DNA to find the sequence of protein. DNA is found naturally in nucleus. Value is to separate DNA from everything else.
Lab 4i
Pouring agarose gels
8% agarose in 1X TAE(Tris- acetate- EDTA)
Tris- maintains pH, buffer
CH3C00-(acetate) - keeps DNA from clumping together
EDTA- prevents DNase
Procedure
1. Make 500 mL 1X TAE + 50 mL, 8% agarose
Use 500 mL g.L and 250 mL erlenmeyer flask and 25 mL pipet/ red filter
a. Add agarose to 100mL 1X TAE in flask.
b. Heat to boil and dissolve- heat swirl -heat-swirl until clear
c. Let cool until you can touch flask for a few seconds.
d. Poor in prepared mold and let cool.
50 mL 0.8% agarose in 1X TAE
% by volume
0.8%= of 50 mL
0.008 x 50 = 0.4 grams agarose in 50 mL 1X TAE
All in erlenmeyer flask
1X TAE
Make from 40X stock
C1V1=C2V2
V1 = C2V2/C1
C1= stock concentration
V1 = 12.5 mL volume stock
C2= final concentration
V2= final volume
V1= (1)(500)/(40)= 12.5 mL of 40X TAE and H2O up to 500 mL (qs to 500 mL with H2O)
Lab 4j
1. Remove tape from gel, place in gel tank
2. Pour TAE over gel until covered, gently remove combs.
3. Prepare samples
20 mL DNA and 4 mL 6X loading dye
spin 2 seconds in mini centrifuge
4. Load samples onto gel
5. Put cover on gel tank, plus to power supply
6. Run at 110V for about 45 minutes
7. Stain for several hours with EtBr (Ethidium Bromide) ; rise and observe with light
Loading dye
-Dyes to track gel process (runs in front of sample)
- Glycerol- to make samples sink into well
Analytical balance
Tabletop milligram balance
7.6 cm x 7.6 cm weigh paper
3.5 inches x 3.5 inches weigh boat
Lab scoops
Sodium chloride
Tubes, 15 mL, capped
Tube racks for 15 mL tubes
TRIS
EDTA, disodium salt
Bottle, 125 mL
Graduated cylinder, 100 mL
Ph paper
Hydrochloric acid
Sodium hydroxide
Glass rods
50 mL beakers
Salmon sperm sample
2mL pipet
Micropipet, P-1000
95% Ethanol
Sharpie
Plastic beaker 1L tripour
40x TAE buffer concentrate
600 mL beaker
Agarose
250 mL Media bottle
Microwave Oven
Hot hands protector
Horizontal gel box
65 degree Celsius water bath
Prepared agarose gel
Tube rack for 1.7 mL tubes
Reaction tubes, 1.7 mL
DNA samples
Yeast DNA 50 micrometers (g)/micrometer(liter) and loading dye
pBR322 50 micrometers (g)/micrometer(liter) and dye
Lambda 50 micrometers (g)/micrometer(liter) and loading dye
Other loading samples and dye
Gel loading dye 10X or 6X
Micropipet, P-10
Micropipet, P-100
Micropipet tips for P-100
Microcentrifuge
Lambda/ HimIII, 50 micrometers (g)/micrometer(liter) and dye
power supply
Ethidium bromide, 0.5 micrometer(g)/ mL
Gel photo imaging system
Thermal paper
Thermal paper
Large gloves
Safety plastic glasses
5.5 inches x 5.5 inches weigh boat
Lab 4a and 4b
Why NaCl? Na+ bind DNA so it can clump together and can precipitate
Precipitation- taking something out of a solution
DNases- breaks down DNA
Why Tris? buffer, maintains pH
why EDTA? prevents DNA braked down negative binds ca++ and mg++ co-factors enzymes. Prevents DNases activity
Purpose:
to make 10 ml of 5m NaCl
100mL of 10mM Tris, 1mM EDTA
M= mole/L
mM= 1/1000 mole/L
1 mole= 6.02 x 10^23 particles
Molarity Calculations
g substance needed=(M)(L)(g/mol)
Solution 1:
5M NaCl = 10mL
1. 10mL 5M NaCl
(5M) (0.01L) (58.44 g/mol) = 2.92 g NaCl NaCl-formula weight = 58.44 g/mol
2. TE
Tris (0.01M)(0.1L)(157.6 g/mol) = 0.158 g Tris
EDTA (0.001M)(0.1L)(372.24 g/mol) = 0.0372 g EDTA
Label
What it is- NaCl
Concentration- 5M desired ph 7.5-8.5 current ph is 6.3-6.5 so need to add base(NaCl)
date- 10-9-14
initials- NR, ZM, JM, RS,MI
period- 2/3
C1V1 (stock solution concentration)=C2V2 (final desired concentration)= final desired volume
C1= volume of stock solution to use V1= C2V2/C1
C=4 mg/ml
V1= ?
C2= 2 mg/ml
V2= 2 mL
Final ph 7.7
Purpose 4b was to spool DNA to extract from liquid.
1. Dilute DNA w/TE -1 mL DNA
1m beaker,observe - 1 mL TE
2. Add NaCl
3. Add 4 mL EtoH(trickle down side) observe
4. Spool DNA
5. Put DNA into new tube w/ 2 mL fresh Te and Label:
Observations
1. The substance is transparent and somewhat congealed.
2. The substance is still clear but there is a different layer and the DNA is showing.
3. The DNA is dense and is more right than the rest of the substance.
4. DNA was kind of viscus and goopy. Me and my partner had similar results to everyone in class.
We left no DNA in beaker. You need to know howto extract DNA so you can study it by finding sequence of DNA to find the sequence of protein. DNA is found naturally in nucleus. Value is to separate DNA from everything else.
Lab 4i
Pouring agarose gels
8% agarose in 1X TAE(Tris- acetate- EDTA)
Tris- maintains pH, buffer
CH3C00-(acetate) - keeps DNA from clumping together
EDTA- prevents DNase
Procedure
1. Make 500 mL 1X TAE + 50 mL, 8% agarose
Use 500 mL g.L and 250 mL erlenmeyer flask and 25 mL pipet/ red filter
a. Add agarose to 100mL 1X TAE in flask.
b. Heat to boil and dissolve- heat swirl -heat-swirl until clear
c. Let cool until you can touch flask for a few seconds.
d. Poor in prepared mold and let cool.
50 mL 0.8% agarose in 1X TAE
% by volume
0.8%= of 50 mL
0.008 x 50 = 0.4 grams agarose in 50 mL 1X TAE
All in erlenmeyer flask
1X TAE
Make from 40X stock
C1V1=C2V2
V1 = C2V2/C1
C1= stock concentration
V1 = 12.5 mL volume stock
C2= final concentration
V2= final volume
V1= (1)(500)/(40)= 12.5 mL of 40X TAE and H2O up to 500 mL (qs to 500 mL with H2O)
Lab 4j
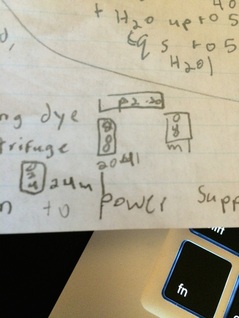
1. Remove tape from gel, place in gel tank
2. Pour TAE over gel until covered, gently remove combs.
3. Prepare samples
20 mL DNA and 4 mL 6X loading dye
spin 2 seconds in mini centrifuge
4. Load samples onto gel
5. Put cover on gel tank, plus to power supply
6. Run at 110V for about 45 minutes
7. Stain for several hours with EtBr (Ethidium Bromide) ; rise and observe with light
Loading dye
-Dyes to track gel process (runs in front of sample)
- Glycerol- to make samples sink into well
|
|
Conclusion
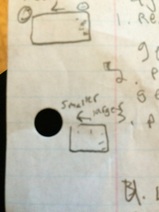
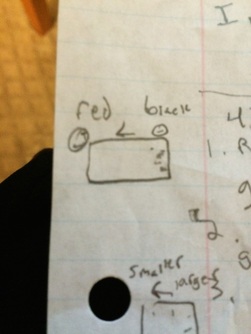
When we did this experiment it didn't go as planned. There was no movement of the DNA when we casted the light on it and the reasons are:
- The staining time was too short. This is probably not the reason but it is possible.
- The solution didn't respond (mix DNA in).
- The solution was made incorrectly. Not likely because that means every group must have did it wrong.
- DNA ran off the gel- not likely but it could have diffused out of gel.
- EtBr could be bad.
We could do tests that could help us figure out what went wrong.
1. Dot blot test with CtBr solution and drop DNA on that. It should become orange.
2. Make EtBr again (fresh) and re-stain gels.
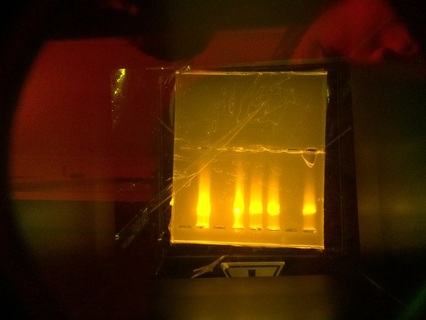
After we re-stained the solution and the DNA appeared. The left gel at the bottom of the page is the redid gel and DNA molecules had been moved.
Reflection
The end goal was not achieved, but my group and I worked pretty good together. We could have been more efficient because there were a few of the group members sitting down and not fully participating, but we still finished. From my knowledge we didm't make any mistakes because we did exactly what the book and teacher told us to. We can avoid this by reading the book and ask lots of questions to the teacher (which is why we didn't make any mistakes in this lab). I Like to think my skills at pipetting are very good because I make almost the exact amount of solution we always need. I think making solutions and adjusting pH are good because I take my time and double check my measurements each time. Also I never cross-contaminate the solutions which can cause this experiment to go wrong. Practice never hurts though because as the time goes on we will be more efficient at each step. This was a very interesting lab because even though the lab didn't work we still made a really cool congealed substance and we got to use equipment that we didn't even know that existed.
When we did this experiment it didn't go as planned. There was no movement of the DNA when we casted the light on it and the reasons are:
- The staining time was too short. This is probably not the reason but it is possible.
- The solution didn't respond (mix DNA in).
- The solution was made incorrectly. Not likely because that means every group must have did it wrong.
- DNA ran off the gel- not likely but it could have diffused out of gel.
- EtBr could be bad.
We could do tests that could help us figure out what went wrong.
1. Dot blot test with CtBr solution and drop DNA on that. It should become orange.
2. Make EtBr again (fresh) and re-stain gels.
After we re-stained the solution and the DNA appeared. The left gel at the bottom of the page is the redid gel and DNA molecules had been moved.
Reflection
The end goal was not achieved, but my group and I worked pretty good together. We could have been more efficient because there were a few of the group members sitting down and not fully participating, but we still finished. From my knowledge we didm't make any mistakes because we did exactly what the book and teacher told us to. We can avoid this by reading the book and ask lots of questions to the teacher (which is why we didn't make any mistakes in this lab). I Like to think my skills at pipetting are very good because I make almost the exact amount of solution we always need. I think making solutions and adjusting pH are good because I take my time and double check my measurements each time. Also I never cross-contaminate the solutions which can cause this experiment to go wrong. Practice never hurts though because as the time goes on we will be more efficient at each step. This was a very interesting lab because even though the lab didn't work we still made a really cool congealed substance and we got to use equipment that we didn't even know that existed.
|

|
